Adjusting for batch effects with linear models
Introduction
To illustrate how we can adjust for batch effects using statistical methods, we will create a data example in which the outcome of interest is confounded with batch but not completely. We will also select a outcome for which we have an expectation of what genes should be diferentially expressed. Namely, we make sex the outcome of interest and expect genes on the Y chromosome to be diferentially expressed. Note that we may also see genes from the X chromosome as differentially expressed as some escape X inactivation. This example dataset is here
library(GSE5859Subset)
data(GSE5859Subset)
To illustrate the confounding we will pick some genes to show in a heatmap plot. We pick all Y chromosome genes, some genes that we see correlate with batch, and then some randomly selected genes.
library(rafalib)
library(genefilter)
batch <- factor(format(sampleInfo$date,"%m"))
chr <- geneAnnotation$CHR
tt<-rowttests(geneExpression,batch)
ind1 <- which(chr=="chrY") ##real differences
ind2 <- setdiff(c(order(tt$dm)[1:25],order(-tt$dm)[1:25]),ind1)
set.seed(1)
ind0 <- setdiff(sample(seq(along=tt$dm),50),c(ind2,ind1))
geneindex<-c(ind2,ind0,ind1)
mat<-geneExpression[geneindex,]
mat <- mat -rowMeans(mat)
icolors <- colorRampPalette(rev(brewer.pal(11,"RdYlBu")))(100)
mypar(1,1)
image(t(mat),xaxt="n",yaxt="n",col=icolors)
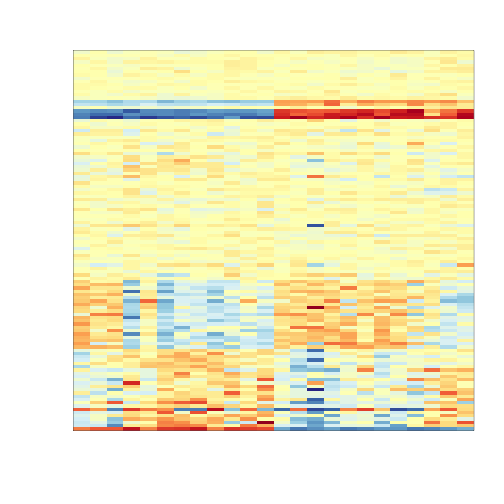
In what follows we will imitate the typical analysis we would do in practice. We will act as if we don’t know which genes are supposed to be differentially expressed between males and females.
Exploratory data analysis for evaluation
Another reason we are using the dataset described above for illustrating different approaches is that in that we actually reasonable idea of what to expect. Autosomal (not on chrX or chrY) genes on the list are likely false positives and chr Y are likely true positives. Chr X genes could go either way. This gives us the opportunity to compare different procedures. In practice we rarely know the “truth” these evaluations are not possible. Simulations are therefore commonly used for evaluation purposes: we know the truth because we construct the data. But simulations are at risk of not capturing all the nuances of real experimental data. This dataset is an experimental dataset
Through the next sections we will use the histogram p-values to evaluate the specificity (low false positive rates) of the batch adjustment procedures presented here. Because the autosomal genes are not expected to be differentially expressed we should see a a flat p-value histogram. To evaluate sensitivity (low false negative rates) we will report the number of the number of reported genes on chrX and chrY. Here are the results when we don’t adjust and report genes with q-values smaller than 0.1. We also include a volcano plot with a horizontal dashed line separating genes called significant and those don’t and color used to highlight chrX and chrY genes.
library(qvalue)
res <- rowttests(geneExpression,as.factor( sampleInfo$group ))
mypar2(1,2)
hist(res$p.value[which(!chr%in%c("chrX","chrY") )],main="",ylim=c(0,1300))
plot(res$dm,-log10(res$p.value))
points(res$dm[which(chr=="chrX")],-log10(res$p.value[which(chr=="chrX")]),col=1,pch=16)
points(res$dm[which(chr=="chrY")],-log10(res$p.value[which(chr=="chrY")]),col=2,pch=16,xlab="Effect size",ylab="-log10(p-value)")
legend("bottomright",c("chrX","chrY"),col=1:2,pch=16)
qvals <- qvalue(res$p.value)$qvalue
index <- which(qvals<0.1)
abline(h=-log10(max(res$p.value[index])))
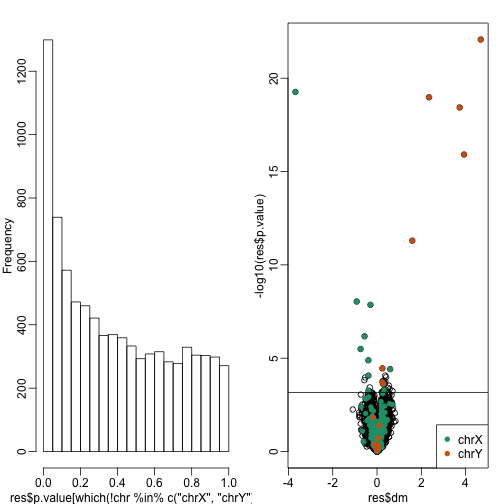
cat("Total genes with q-value < 0.1:",length(index))
## Total genes with q-value < 0.1: 41
cat("Number of selected genes on chrY:", sum(chr[index]=="chrY",na.rm=TRUE))
## Number of selected genes on chrY: 8
cat("Number of selected genes on chrX:", sum(chr[index]=="chrX",na.rm=TRUE))
## Number of selected genes on chrX: 11
Note that the histogram is not flat. Instead, low p-values are over-represented. More than half of the genes on the final list are autosomal.
Adjusting with liner models
We have already noted that processing date has an effect on gene expression thus we will try to adjust for thi s by including it a model. When we perform a t-test comparing the two groups it is equivalent to fitting the following linear model:
to each gene with if subject is female and 0 otherwise. Note that represent the estimated difference for gene and represents the withing group variation. So what is the problem?
The theory we described the linear models chapter assumes that the error terms are independent. In the case we know that this is not the case for all genes because we know the error terms from October will be more alike to each other than to the June error terms. We can adjust for this by including a term in that models this effect:
Here if sample was processed in October and 0 otherwise and is the month effect for gene . Note that this an example of how linear models give us much more flexible than procedures such as the t-test.
We construct a model matrix that includes batch.
sex <- sampleInfo$group
X <- model.matrix(~sex+batch)
Now we can fit a model for each gene. For example note the difference between the original model and one that adjusted for batch:
j <- 7635
y <- geneExpression[j,]
X0 <- model.matrix(~sex)
fit <- lm(y~X0-1)
summary(fit)$coef
## Estimate Std. Error t value Pr(>|t|)
## X0(Intercept) 6.9555747 0.2166035 32.112008 5.611901e-20
## X0sex -0.6556865 0.3063237 -2.140502 4.365102e-02
X <- model.matrix(~sex+batch)
fit <- lm(y~X)
summary(fit)$coef
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 7.26329968 0.1605560 45.2384140 2.036006e-22
## Xsex -0.04023663 0.2427379 -0.1657616 8.699300e-01
## Xbatch10 -1.23089977 0.2427379 -5.0709009 5.070727e-05
We then fit this new model for each gene. For example we can use sapply to recover the estimated coefficient and p-value in the following way:
res <- t( sapply(1:nrow(geneExpression),function(j){
y <- geneExpression[j,]
fit <- lm(y~X-1)
summary(fit)$coef[2,c(1,4)]
} ) )
##turn into data.frame so we can use the same code for plots as above
res <- data.frame(res)
names(res) <- c("dm","p.value")
mypar2(1,2)
hist(res$p.value[which(!chr%in%c("chrX","chrY") )],main="",ylim=c(0,1300))
plot(res$dm,-log10(res$p.value))
points(res$dm[which(chr=="chrX")],-log10(res$p.value[which(chr=="chrX")]),col=1,pch=16)
points(res$dm[which(chr=="chrY")],-log10(res$p.value[which(chr=="chrY")]),col=2,pch=16,xlab="Effect size",ylab="-log10(p-value)")
legend("bottomright",c("chrX","chrY"),col=1:2,pch=16)
qvals <- qvalue(res$p.value)$qvalue
index <- which(qvals<0.1)
abline(h=-log10(max(res$p.value[index])))
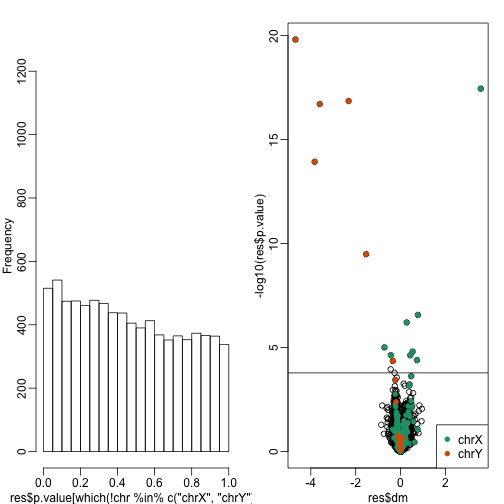
cat("Total genes with q-value < 0.1:",length(index))
## Total genes with q-value < 0.1: 16
cat("Number of selected genes on chrY:", sum(chr[index]=="chrY",na.rm=TRUE))
## Number of selected genes on chrY: 6
cat("Number of selected genes on chrX:", sum(chr[index]=="chrX",na.rm=TRUE))
## Number of selected genes on chrX: 8
Note the great improvement in specificity (less false positives) without much loss in sensitivity (we still find many chrY genes). However, we stil see some bias in the histogram. In the following sections we will see that month does not perfectly account for the batch effect and better estimates are possible.
A note on computing efficiency
Note that in the code above the design matrix does not change within the iterations we are computing over and over and applying it to each gene. Instead we can perform this calculation in one matrix algebra calculation by computing it once and then obtaining all the betas by multiplying with the columns of representing genes in this case. The limma package has an implementation of this idea (using the QR decomposition). Note how much faster this is:
library(limma)
X <- model.matrix(~sex+batch)
fit <- lmFit(geneExpression,X)
The estimated regression coefficients for each gene are obtained like this:
dim( fit$coef)
## [1] 8793 3
Note we have one estimate for each gene. To obtain p-values for one of these we have to construct the ratios:
k <- 2 ##second coef
ses <- fit$stdev.unscaled[,k]*fit$sigma
ttest <- fit$coef[,k]/ses
pvals <- 2*pt(-abs(ttest),fit$df)
We will cover limma in more detail in a later section.