Sharded GRanges: a hybrid in/out of memory strategy for large sets of ranges
Introduction
We’ve looked at a number of approaches to working with data external to R:
- HDF5, which manages groups of multidimensional arrays on disk
- sqlite, a zero-configuration relational database
- tabix, a simple approach to indexing records on genomic coordinates
Here I want to describe an approach that seems useful for millions of ranges annotated in the course of searching for variants that affect gene expression at the population level. The approach is based on a concept of storing data in “shards”, homogeneous small fragments that can be quickly loaded and unloaded, discoverable by index and traversable in parallel.
Motivation: An integrative view of associations in GEUVADIS
The GEUVADIS study is an intensive multiomic study of gene expression in multiple populations. We want to make use of the data from this study to investigate variants affecting genes of interest, with one tool an interactive graphical utility illustrated in the video:
library(ph525x)
ggshot()
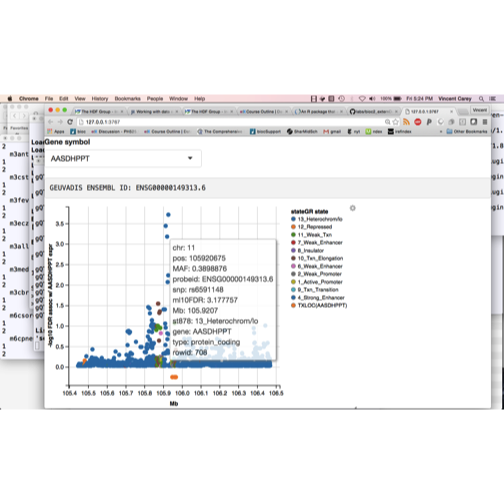
We want to be able to select genes by symbol and explore names and epigenetic contexts of variants whose content is associated with expression variation. It is useful to have the variants annotated using GRanges, but a very large GRanges object (there are hundreds of millions of SNP-gene associations recorded) can be unwieldy. Solutions using RDBMS or HDF5 may be viable but more infrastructure for rapidly searching such stores using genomic coordinates, and for converting query results to GRanges will be needed.
BatchJobs was used to generate the association tests, and it produces an organized system of “sharded” GRanges recording the associations along with metadata about the associated features. This system can be stored in a package, exemplified by geuvStore.
A quick look at geuvStore
The association test results are organized using a BatchJobs registry that is wrapped in an S4 class called ciseStore.
library(geuvStore2)
m = makeGeuvStore2()
class(m)
## [1] "ciseStore"
## attr(,"package")
## [1] "gQTLBase"
m
## ciseStore instance with 160 completed jobs.
## excerpt from job 1 :
## GRanges object with 1 range and 14 metadata columns:
## seqnames ranges strand | paramRangeID REF
## <Rle> <IRanges> <Rle> | <factor> <DNAStringSet>
## [1] 1 [526736, 526736] * | ENSG00000215915.5 C
## ALT chisq permScore_1 permScore_2 permScore_3
## <CharacterList> <numeric> <numeric> <numeric> <numeric>
## [1] G 2.463829 3.145667 0.4092251 0.1571743
## permScore_4 permScore_5 permScore_6 snp MAF
## <numeric> <numeric> <numeric> <character> <numeric>
## [1] 0.02981471 0.1648088 0.0123114 rs28863004 0.09101124
## probeid mindist
## <character> <numeric>
## [1] ENSG00000215915.5 858333
## -------
## seqinfo: 86 sequences from hg19 genome
The show method for m probes into the store and retrieves one record from one GRanges instance.
Scalable traversal
The traversal of all GRanges available in this selection is governed by foreach loops.
library(gQTLBase)
ut1 = system.time(l1 <- storeApply(m, length))
## Warning: executing %dopar% sequentially: no parallel backend registered
ut1
## user system elapsed
## 18.296 0.856 21.051
library(doParallel)
registerDoParallel(cores=2)
ut2 = system.time(l2 <- storeApply(m, length))
ut2
## user system elapsed
## 10.028 1.694 14.087
print(sum(unlist(l2)))
## [1] 6183186
all.equal(unlist(l1), unlist(l2))
## [1] TRUE
We see that doubling the number of processors reduces the time required to get the length of each component of the archive. With large numbers of cores, we can quickly assemble information about many variants.
Scalable histogram construction
When the histogram bins are fixed, divide and conquer can be used to assemble a histogram in parallel over many chunks.
registerDoParallel(cores=1)
system.time(ll <- storeToHist(m, getter=function(x)log(mcols(x)$chisq+1), breaks=c(0,seq(.1,5,.1),10)))
## user system elapsed
## 25.243 1.010 27.337
registerDoParallel(cores=2)
system.time(ll <- storeToHist(m, getter=function(x)log(mcols(x)$chisq+1), breaks=c(0,seq(.1,5,.1),10)))
## user system elapsed
## 31.753 4.979 16.576
Indexing for targeted retrievals
The ciseStore class includes two maps: one from range to shard number, another from gene identifier to shard number. This allows rapid retrievals.
myr = GRanges(2, IRanges(1975.7e5, width=50000))
extractByRanges(m, myr)
## GRanges object with 190 ranges and 15 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## [1] 2 [197570297, 197570297] * | ENSG00000247626.2
## [2] 2 [197570357, 197570357] * | ENSG00000247626.2
## [3] 2 [197570746, 197570746] * | ENSG00000247626.2
## [4] 2 [197570827, 197570827] * | ENSG00000247626.2
## [5] 2 [197570836, 197570836] * | ENSG00000247626.2
## ... ... ... ... . ...
## [186] 2 [197619213, 197619213] * | ENSG00000081320.5
## [187] 2 [197619564, 197619564] * | ENSG00000081320.5
## [188] 2 [197619731, 197619731] * | ENSG00000081320.5
## [189] 2 [197619765, 197619765] * | ENSG00000081320.5
## [190] 2 [197619940, 197619940] * | ENSG00000081320.5
## REF ALT chisq permScore_1
## <DNAStringSet> <CharacterList> <numeric> <numeric>
## [1] A G 2.03938471 2.227660978
## [2] A G 0.00219115 2.308633433
## [3] C T 0.02237336 0.007156323
## [4] C A 3.19288246 2.378335741
## [5] A G 3.35555840 2.028726357
## ... ... ... ... ...
## [186] A G 7.463969e-01 2.0296567
## [187] T C 9.167750e-02 0.9853224
## [188] T G 1.616341e-04 3.4520666
## [189] T G 1.546168e+01 0.3229887
## [190] C G 1.546168e+01 0.3229887
## permScore_2 permScore_3 permScore_4 permScore_5 permScore_6
## <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 5.59899744 0.6579064 0.9328838 4.567436 1.981865134
## [2] 2.93511183 1.7106360 0.2018579 1.020219 0.022856700
## [3] 0.07385741 0.2701249 0.8719252 4.967324 0.007260719
## [4] 0.44403377 1.0562575 0.1413580 1.118612 0.371785413
## [5] 0.68500429 0.8265180 0.2048171 1.435278 0.143369095
## ... ... ... ... ... ...
## [186] 2.4329376 0.0439362 0.3414404 4.2774530 0.1259558
## [187] 1.4412183 0.5267052 0.6940248 1.2478614 1.3928722
## [188] 1.7408850 0.0416719 0.4461964 0.1202545 1.0351946
## [189] 0.7242746 2.2331430 0.2837090 1.1147593 0.4177635
## [190] 0.7242746 2.2331430 0.2837090 1.1147593 0.4177635
## snp MAF probeid mindist jobid
## <character> <numeric> <character> <numeric> <integer>
## [1] rs142213149 0.02921348 ENSG00000247626.2 999790 95
## [2] rs146312688 0.01797753 ENSG00000247626.2 999730 95
## [3] rs4850726 0.22471910 ENSG00000247626.2 999341 95
## [4] rs9750402 0.08314607 ENSG00000247626.2 999260 95
## [5] rs9752394 0.08426966 ENSG00000247626.2 999251 95
## ... ... ... ... ... ...
## [186] rs62185596 0.215730337 ENSG00000081320.5 577986 101
## [187] rs7583693 0.298876404 ENSG00000081320.5 578337 101
## [188] rs145286171 0.030337079 ENSG00000081320.5 578504 101
## [189] rs147575472 0.003370787 ENSG00000081320.5 578538 101
## [190] rs140304200 0.003370787 ENSG00000081320.5 578713 101
## -------
## seqinfo: 86 sequences from hg19 genome
Conclusions
geuvStore2 is a complex architecture that aims to provide a partly baked representation of quantities from genome-scale surveys that can be scalably surveyed and integrated. This is accomplished by keeping ranges for association scores and metadata in small sharded GRanges with some simple indexes, retrieval utilities, and with support for parallelized traversal and summary. It would be very nice to achieve these aims with a more homogeneous underlying architecture such as HDF5, and this may be possible as file-backed SummarizedExperiments come on line.