Computing with chromosomes and variants
Overview
We will consider how to do various very high-level tasks with chromosomes and variants in Bioconductor.
- listing packages representing reference builds for humans and model organisms
- acquiring human reference genome sequence
- finding views of genes as sequences
- examining the dbSNP catalog of small variants in populations of human genomes
- examining the NHGRI GWAS catalog of associations between variants and phenotypes
BSgenome and available genomes
library(BSgenome)
head(available.genomes()) # requires internet access
## [1] "BSgenome.Alyrata.JGI.v1"
## [2] "BSgenome.Amellifera.BeeBase.assembly4"
## [3] "BSgenome.Amellifera.UCSC.apiMel2"
## [4] "BSgenome.Amellifera.UCSC.apiMel2.masked"
## [5] "BSgenome.Athaliana.TAIR.04232008"
## [6] "BSgenome.Athaliana.TAIR.TAIR9"
grep("Hsapiens", available.genomes(), value=TRUE)
## [1] "BSgenome.Hsapiens.NCBI.GRCh38"
## [2] "BSgenome.Hsapiens.UCSC.hg17"
## [3] "BSgenome.Hsapiens.UCSC.hg17.masked"
## [4] "BSgenome.Hsapiens.UCSC.hg18"
## [5] "BSgenome.Hsapiens.UCSC.hg18.masked"
## [6] "BSgenome.Hsapiens.UCSC.hg19"
## [7] "BSgenome.Hsapiens.UCSC.hg19.masked"
The human reference sequence, build hg19; gene sequence
library(BSgenome.Hsapiens.UCSC.hg19)
Hsapiens
## Human genome
## |
## | organism: Homo sapiens (Human)
## | provider: UCSC
## | provider version: hg19
## | release date: Feb. 2009
## | release name: Genome Reference Consortium GRCh37
## | 93 sequences:
## | chr1 chr2 chr3
## | chr4 chr5 chr6
## | chr7 chr8 chr9
## | chr10 chr11 chr12
## | chr13 chr14 chr15
## | ... ... ...
## | chrUn_gl000235 chrUn_gl000236 chrUn_gl000237
## | chrUn_gl000238 chrUn_gl000239 chrUn_gl000240
## | chrUn_gl000241 chrUn_gl000242 chrUn_gl000243
## | chrUn_gl000244 chrUn_gl000245 chrUn_gl000246
## | chrUn_gl000247 chrUn_gl000248 chrUn_gl000249
## | (use 'seqnames()' to see all the sequence names, use the '$' or '[['
## | operator to access a given sequence)
c17 = Hsapiens$chr17
c17
## 81195210-letter "DNAString" instance
## seq: AAGCTTCTCACCCTGTTCCTGCATAGATAATTGC...GGTGTGGGTGTGGTGTGTGGGTGTGGGTGTGGT
The class of c17 is DNAString. This is a full in-memory representation of all the bases of the chromosome. We can work with substructures of interest without duplicating the contents of memory devoted to the sequence. We’ll obtain a view of coding sequences of genes on chromosome 17. To do this we will employ a special transcript database structure.
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
txdb
## TxDb object:
## | Db type: TxDb
## | Supporting package: GenomicFeatures
## | Data source: UCSC
## | Genome: hg19
## | Organism: Homo sapiens
## | UCSC Table: knownGene
## | Resource URL: http://genome.ucsc.edu/
## | Type of Gene ID: Entrez Gene ID
## | Full dataset: yes
## | miRBase build ID: GRCh37
## | transcript_nrow: 82960
## | exon_nrow: 289969
## | cds_nrow: 237533
## | Db created by: GenomicFeatures package from Bioconductor
## | Creation time: 2014-09-26 11:16:12 -0700 (Fri, 26 Sep 2014)
## | GenomicFeatures version at creation time: 1.17.17
## | RSQLite version at creation time: 0.11.4
## | DBSCHEMAVERSION: 1.0
We are only interested in information on chr17 at the moment. We establish chr17 as the active sequence in this transcript database
tmp = isActiveSeq(txdb)
tmp[] = FALSE # turn all off
tmp[17] = TRUE # turn 17 on
isActiveSeq(txdb) = tmp
g17 = genes(txdb)
g17
## GRanges object with 1357 ranges and 1 metadata column:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <character>
## 100124536 chr17 [65736786, 65736917] + | 100124536
## 100126313 chr17 [11985216, 11985313] + | 100126313
## 100126356 chr17 [29902430, 29902540] + | 100126356
## 100128288 chr17 [ 8261731, 8263859] - | 100128288
## 100128977 chr17 [43920722, 43972879] - | 100128977
## ... ... ... ... ... ...
## 9931 chr17 [65066554, 65241319] - | 9931
## 9953 chr17 [14204506, 14249492] + | 9953
## 9955 chr17 [13399006, 13505244] - | 9955
## 996 chr17 [45195311, 45266665] - | 996
## 9969 chr17 [60019966, 60142643] - | 9969
## -------
## seqinfo: 1 sequence from hg19 genome
Now we make a structure that has addresses and sequences of genes.
gs17 = getSeq(Hsapiens, g17)
gs17
## A DNAStringSet instance of length 1357
## width seq names
## [1] 132 CCTCCTACAAAGGCATGT...AACCATCCCACATAGAA 100124536
## [2] 98 TTGGGCAAGGTGCGGGGC...CTCAACCTTACTCGGTC 100126313
## [3] 111 AGAGTGTTCAAGGACAGC...TTGCAGTGTGCATCGGG 100126356
## [4] 2129 TTTTTTAGTTCCTGGTTC...GAGCAAGACTCTGTCTC 100128288
## [5] 52158 GCGGCCGCCGAGTCCGTC...ATTAGGGCCCACCCTAA 100128977
## ... ... ...
## [1353] 174766 ATGCTTTGTGGGGACGTT...CTTTTTAATAATTTGGA 9931
## [1354] 44987 GTGGCCAGGGCGCGAGAG...ACAGCCCTAGGGTTCTG 9953
## [1355] 106239 CAGCGGCGGCCCAGGAGG...TAAAAAAATAAATCTTG 9955
## [1356] 71355 AATCGCTCGGCCTCCCCC...GGATATGGAAAATCCAA 996
## [1357] 122678 GTTTCTCTCTCTGGTCGG...TATTCATGAAGCCTTCA 9969
In the next version of Bioconductor this can be accomplished somewhat more efficiently using “Views()”.
dbSNP
We have an image of the dbSNP variant catalog for hg19. The information retained is limited to the dbSNP identifier, chromosome location, and variant content.
library(SNPlocs.Hsapiens.dbSNP.20120608)
sl17 = getSNPlocs("ch17", as.GRanges=TRUE)
sl17
## GRanges object with 1247508 ranges and 2 metadata columns:
## seqnames ranges strand | RefSNP_id
## <Rle> <IRanges> <Rle> | <character>
## [1] ch17 [ 56, 56] + | 145615430
## [2] ch17 [ 78, 78] + | 148170422
## [3] ch17 [ 80, 80] + | 183779916
## [4] ch17 [174, 174] + | 188505217
## [5] ch17 [293, 293] + | 9747578
## ... ... ... ... ... ...
## [1247504] ch17 [81190378, 81190378] + | 71264801
## [1247505] ch17 [81190400, 81190400] + | 74838487
## [1247506] ch17 [81193098, 81193098] + | 77334326
## [1247507] ch17 [81194235, 81194235] + | 182545986
## [1247508] ch17 [81194907, 81194907] + | 187112992
## alleles_as_ambig
## <character>
## [1] Y
## [2] S
## [3] R
## [4] R
## [5] R
## ... ...
## [1247504] R
## [1247505] R
## [1247506] R
## [1247507] Y
## [1247508] K
## -------
## seqinfo: 25 sequences (1 circular) from GRCh37.p5 genome
The allele codes are translated by the IUPAC map.
IUPAC_CODE_MAP
## A C G T M R W S Y K
## "A" "C" "G" "T" "AC" "AG" "AT" "CG" "CT" "GT"
## V H D B N
## "ACG" "ACT" "AGT" "CGT" "ACGT"
GWAS catalog
National Human Genome Research Institute maintains a listing of genetic association studies that have found significant associations between DNA variants and major phenotypes and diseases. Inclusion in the catalog requires that the findings be replicated in an independent population.
library(gwascat)
data(gwrngs19) # for hg19
gwrngs19
## gwasloc instance with 17254 records and 35 attributes per record.
## Extracted: Mon Sep 8 13:08:13 2014
## Genome: hg19
## Excerpt:
## GRanges object with 5 ranges and 3 metadata columns:
## seqnames ranges strand | Disease.Trait
## <Rle> <IRanges> <Rle> | <character>
## 1 chr19 [ 7739177, 7739177] * | Resistin levels
## 2 chr6 [ 32626601, 32626601] * | Asthma and hay fever
## 3 chr4 [ 38799710, 38799710] * | Asthma and hay fever
## 4 chr5 [110467499, 110467499] * | Asthma and hay fever
## 5 chr2 [102966549, 102966549] * | Asthma and hay fever
## SNPs p.Value
## <character> <numeric>
## 1 rs1423096 1e-07
## 2 rs9273373 4e-14
## 3 rs4833095 5e-12
## 4 rs1438673 3e-11
## 5 rs10197862 4e-11
## -------
## seqinfo: 23 sequences from hg19 genome
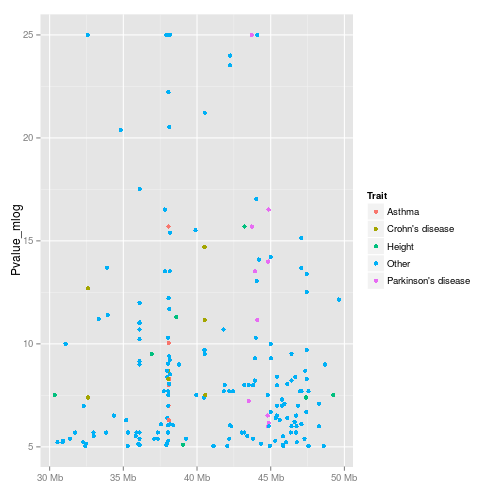
A simple display of associations and phenotypes is available
with the traitsManh function.
example(traitsManh)
##
## trtsMn> # do a p-value truncation if you want to reduce compression
## trtsMn> #gwascat:::.onAttach("A", "gwascat")
## trtsMn> data(gwrngs19)
##
## trtsMn> traitsManh(gwrngs19)
## Loading required package: ggbio
## Loading required package: ggplot2
## Need specific help about ggbio? try mailing
## the maintainer or visit http://tengfei.github.com/ggbio/
##
## Attaching package: 'ggbio'
##
## The following objects are masked from 'package:ggplot2':
##
## geom_bar, geom_rect, geom_segment, ggsave, stat_bin,
## stat_identity, xlim