Reading in microarray data
Affymterix CEL files
We start by reading in the sample information table. This is usually created by the person who performed the experiment.
The raw data files for this lab are in the rawdata repository, available here:
https://github.com/genomicsclass/rawdata
Click Download ZIP in order to download all the files, unzip this file which should result in a rawdata-master folder.
First we save the initial working directory, so we can return to it.
wd <- getwd()
Now we can start reading in the files:
datadir <- "rawdata-master"
basedir <- paste0(datadir, "/celfiles")
setwd(basedir)
library(affy)
## Loading required package: BiocGenerics
## Loading required package: methods
## Loading required package: parallel
##
## Attaching package: 'BiocGenerics'
##
## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB
##
## The following object is masked from 'package:stats':
##
## xtabs
##
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, as.vector, cbind,
## colnames, do.call, duplicated, eval, evalq, Filter, Find, get,
## intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rep.int, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unlist
##
## Loading required package: Biobase
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.
tab <- read.delim("sampleinfo.txt", check.names = FALSE, as.is = TRUE)
rownames(tab) <- tab$filenames
tab
## filenames 37777_at 684_at 1597_at
## 1521a99hpp_av06.CEL.gz 1521a99hpp_av06.CEL.gz 0.00 0.25 0.5
## 1532a99hpp_av04.CEL.gz 1532a99hpp_av04.CEL.gz 0.00 0.25 0.5
## 2353a99hpp_av08.CEL.gz 2353a99hpp_av08.CEL.gz 0.00 0.25 0.5
## 1521b99hpp_av06.CEL.gz 1521b99hpp_av06.CEL.gz 0.25 0.50 1.0
## 1532b99hpp_av04.CEL.gz 1532b99hpp_av04.CEL.gz 0.25 0.50 1.0
## 2353b99hpp_av08r.CEL.gz 2353b99hpp_av08r.CEL.gz 0.25 0.50 1.0
## 38734_at 39058_at 36311_at 36889_at 1024_at
## 1521a99hpp_av06.CEL.gz 1 2 4 8 16
## 1532a99hpp_av04.CEL.gz 1 2 4 8 16
## 2353a99hpp_av08.CEL.gz 1 2 4 8 16
## 1521b99hpp_av06.CEL.gz 2 4 8 16 32
## 1532b99hpp_av04.CEL.gz 2 4 8 16 32
## 2353b99hpp_av08r.CEL.gz 2 4 8 16 32
## 36202_at 36085_at 40322_at 407_at 1091_at 1708_at
## 1521a99hpp_av06.CEL.gz 32 64 128 0.00 512 1024
## 1532a99hpp_av04.CEL.gz 32 64 128 0.00 512 1024
## 2353a99hpp_av08.CEL.gz 32 64 128 0.00 512 1024
## 1521b99hpp_av06.CEL.gz 64 128 256 0.25 1024 0
## 1532b99hpp_av04.CEL.gz 64 128 256 0.25 1024 0
## 2353b99hpp_av08r.CEL.gz 64 128 256 0.25 1024 0
## 33818_at 546_at
## 1521a99hpp_av06.CEL.gz 256 32
## 1532a99hpp_av04.CEL.gz 256 32
## 2353a99hpp_av08.CEL.gz 256 32
## 1521b99hpp_av06.CEL.gz 512 64
## 1532b99hpp_av04.CEL.gz 512 64
## 2353b99hpp_av08r.CEL.gz 512 64
fns <- list.celfiles()
fns
## [1] "1521a99hpp_av06.CEL.gz" "1521b99hpp_av06.CEL.gz"
## [3] "1532a99hpp_av04.CEL.gz" "1532b99hpp_av04.CEL.gz"
## [5] "2353a99hpp_av08.CEL.gz" "2353b99hpp_av08r.CEL.gz"
fns %in% tab[, 1] ##check
## [1] TRUE TRUE TRUE TRUE TRUE TRUE
ab <- ReadAffy(phenoData = tab)
This creates an AffyBatch object which object contains the information you need.
dim(pm(ab))
## Warning: replacing previous import by 'utils::head' when loading 'hgu95acdf'
## Warning: replacing previous import by 'utils::tail' when loading 'hgu95acdf'
##
## [1] 201807 6
dim(pData(ab))
## [1] 6 17
annotation(ab)
## [1] "hgu95a"
Note, this object You can then preprocess RMA
e <- rma(ab)
## Background correcting
## Normalizing
## Calculating Expression
Now we go back to the previous working directory.
setwd(wd)
If you are not interested in probe level data you could can use this function
setwd(basedir)
ejust <- justRMA(filenames = tab[, 1], phenoData = tab)
dim(ejust)
## Features Samples
## 12626 6
##Agilent data
library(limma)
##
## Attaching package: 'limma'
##
## The following object is masked from 'package:BiocGenerics':
##
## plotMA
library(rafalib)
## Loading required package: RColorBrewer
basedir <- paste0(datadir, "/agilent")
setwd(basedir)
targets <- readTargets("TargetBeta7.txt")
RG <- read.maimages(targets$FileName, source = "genepix")
## Warning: Name partially matched in data frame
## Read 6Hs.195.1.gpr
## Read 6Hs.168.gpr
## Read 6Hs.166.gpr
## Read 6Hs.187.1.gpr
## Read 6Hs.194.gpr
## Read 6Hs.243.1.gpr
MA <- MA.RG(RG, bc.method = "none")
mypar(1, 1)
imageplot(MA$M[, 2], RG$printer, zlim = c(-3, 3))
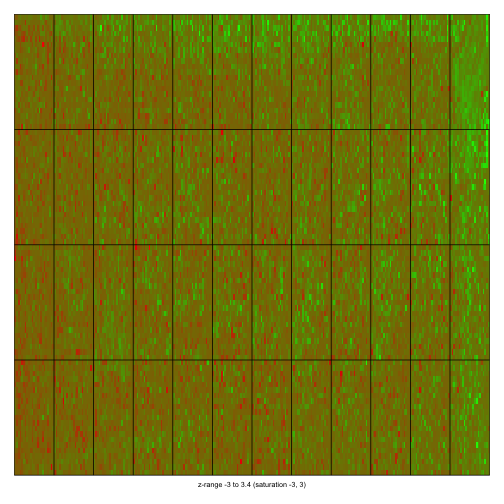
dev.off()
## null device
## 1
Now we go back to the previous working directory.
setwd(wd)
oligo
We can also use oligo to read affy arrays
detach("package:affy")
library(oligo)
## Loading required package: oligoClasses
## Welcome to oligoClasses version 1.26.0
## Loading required package: Biostrings
## Loading required package: IRanges
## Loading required package: XVector
## ===========================================================================
## Welcome to oligo version 1.28.0
## ===========================================================================
##
## Attaching package: 'oligo'
##
## The following object is masked from 'package:limma':
##
## backgroundCorrect
##
## The following object is masked from 'package:BiocGenerics':
##
## normalize
basedir <- paste0(datadir, "/celfiles")
setwd(basedir)
tab <- read.delim("sampleinfo.txt", check.names = FALSE, as.is = TRUE)
fns <- list.celfiles(listGzipped = TRUE)
fns %in% tab[, 1] ##check
## [1] TRUE TRUE TRUE TRUE TRUE TRUE
pd <- as(tab, "AnnotatedDataFrame")
efs <- read.celfiles(filenames = tab[, 1], phenoData = pd, sampleNames = sampleNames(pd))
## Loading required package: pd.hg.u95a
## Loading required package: RSQLite
## Loading required package: DBI
## Platform design info loaded.
## Reading in : 1521a99hpp_av06.CEL.gz
## Reading in : 1532a99hpp_av04.CEL.gz
## Reading in : 2353a99hpp_av08.CEL.gz
## Reading in : 1521b99hpp_av06.CEL.gz
## Reading in : 1532b99hpp_av04.CEL.gz
## Reading in : 2353b99hpp_av08r.CEL.gz
## Warning: 'channel' automatically added to varMetadata in phenoData.
e <- rma(efs)
## Background correcting
## Normalizing
## Calculating Expression