Variant calling and annotation
We start by loading all the libraries we will need for calling and annotating single nucleotide variants (SNVs).
# biocLite('VariantAnnotation') biocLite('VariantTools')
# biocLite('TxDb.Hsapiens.UCSC.hg19.knownGene') biocLite('org.Hs.eg.db')
# biocLite('LungCancerLines') this package is 900 Mb
# biocLite('BSgenome.Hsapiens.UCSC.hg19')
library(VariantTools)
## Loading required package: IRanges
## Loading required package: methods
## Loading required package: BiocGenerics
## Loading required package: parallel
##
## Attaching package: 'BiocGenerics'
##
## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB
##
## The following object is masked from 'package:stats':
##
## xtabs
##
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, as.vector, cbind,
## colnames, do.call, duplicated, eval, evalq, Filter, Find, get,
## intersect, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rep.int, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unlist
##
## Loading required package: GenomicRanges
## Loading required package: GenomeInfoDb
## Loading required package: VariantAnnotation
## Loading required package: Rsamtools
## Loading required package: XVector
## Loading required package: Biostrings
##
## Attaching package: 'VariantAnnotation'
##
## The following object is masked from 'package:base':
##
## tabulate
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
## Loading required package: GenomicFeatures
## Loading required package: AnnotationDbi
## Loading required package: Biobase
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.
library(org.Hs.eg.db)
## Loading required package: DBI
library(LungCancerLines)
library(BSgenome.Hsapiens.UCSC.hg19)
## Loading required package: BSgenome
##
## Attaching package: 'BSgenome'
##
## The following object is masked from 'package:AnnotationDbi':
##
## species
Calling variants
The VariantTools can be used to find variants using a reference genome and a file with aligned reads (BAM file). The reference genome needs to be a GmapGenome, but the GmapGenome function is available to convert any FASTA file to a genome for use with VariantTools.
Here we will examine RNA-Seq reads mapped to the TP53 gene plus 1 Mb flanking sequence.
p53 <- gmapR:::exonsOnTP53Genome("TP53")
genome <- gmapR::TP53Genome()
# ?LungCancerBamFiles
bams <- LungCancerLines::LungCancerBamFiles()
path(bams)
## H1993
## "/usr/local/Cellar/r/3.1.0/R.framework/Versions/3.1/Resources/library/LungCancerLines/extdata/H1993.analyzed.bam"
## H2073
## "/usr/local/Cellar/r/3.1.0/R.framework/Versions/3.1/Resources/library/LungCancerLines/extdata/H2073.analyzed.bam"
bam <- bams$H1993
Again, note that the genome is not hg19.
p53
## GRanges with 21 ranges and 1 metadata column:
## seqnames ranges strand | exon_id
## <Rle> <IRanges> <Rle> | <integer>
## [1] TP53 [1000001, 1000236] - | 224094
## [2] TP53 [1004308, 1004466] - | 224095
## [3] TP53 [1006624, 1007912] - | 224096
## [4] TP53 [1008831, 1008937] - | 224097
## [5] TP53 [1011429, 1011488] - | 224098
## ... ... ... ... ... ...
## [17] TP53 [1014604, 1014844] - | 224110
## [18] TP53 [1014743, 1014841] - | 224111
## [19] TP53 [1014743, 1014844] - | 224112
## [20] TP53 [1015547, 1015649] - | 224113
## [21] TP53 [1025599, 1025772] - | 224114
## ---
## seqlengths:
## TP53
## 2025772
# Bioc 2.13
library(GenomicRanges)
# Bioc 2.14
library(GenomicAlignments)
readGAlignments(bam)
## GAlignments with 4980 alignments and 0 metadata columns:
## seqnames strand cigar qwidth start end
## <Rle> <Rle> <character> <integer> <integer> <integer>
## [1] TP53 + 99M1S 100 237885 237983
## [2] TP53 - 53M107N47M 100 237931 238137
## [3] TP53 + 100M 100 238941 239040
## [4] TP53 - 100M 100 239044 239143
## [5] TP53 + 3S97M 100 258991 259087
## ... ... ... ... ... ... ...
## [4976] TP53 - 74M7376N26M 100 1961426 1968901
## [4977] TP53 + 38M146N62M 100 1961462 1961707
## [4978] TP53 - 41M7156N59M 100 1961679 1968934
## [4979] TP53 + 100M 100 1970185 1970284
## [4980] TP53 - 100M 100 1970259 1970358
## width njunc
## <integer> <integer>
## [1] 99 0
## [2] 207 1
## [3] 100 0
## [4] 100 0
## [5] 97 0
## ... ... ...
## [4976] 7476 1
## [4977] 246 1
## [4978] 7256 1
## [4979] 100 0
## [4980] 100 0
## ---
## seqlengths:
## TP53
## 2025772
We can write out a FASTA file for use in IGV.
x <- as(genome, "DNAStringSet")
library(rtracklayer)
export(x, "genome.fasta")
path(bam)
## [1] "/usr/local/Cellar/r/3.1.0/R.framework/Versions/3.1/Resources/library/LungCancerLines/extdata/H1993.analyzed.bam"
The following call will use a binomial likelihood ratio test to call variants. From the help file:
The test amounts to excluding putative variants with less than ~4% alt frequency. A variant is also required to be represented by at least 2 alt reads.
Regarding base quality, the help says
we typically use 56 for old Illumina, 23 for Sanger/Illumina1.8.
The TallyVariantsParam step takes some time the first time it is run (15 minutes or so).
tally.param <- TallyVariantsParam(genome, high_base_quality = 23L, which = range(p53) +
50000)
called.variants <- callVariants(bam, tally.param)
called.variants
## VRanges with 4 ranges and 15 metadata columns:
## seqnames ranges strand ref alt
## <Rle> <IRanges> <Rle> <character> <characterOrRle>
## [1] TP53 [1003991, 1003991] + T C
## [2] TP53 [1012459, 1012459] + G C
## [3] TP53 [1013114, 1013114] + T C
## [4] TP53 [1014376, 1014376] + G C
## totalDepth refDepth altDepth sampleNames
## <integerOrRle> <integerOrRle> <integerOrRle> <factorOrRle>
## [1] 2 0 2 <NA>
## [2] 8 0 8 <NA>
## [3] 4 0 4 <NA>
## [4] 4 0 4 <NA>
## softFilterMatrix | raw.count raw.count.ref raw.count.total
## <matrix> | <integer> <integer> <integer>
## [1] | 2 0 2
## [2] | 8 0 8
## [3] | 4 0 4
## [4] | 4 0 4
## mean.quality mean.quality.ref count.plus count.plus.ref count.minus
## <numeric> <numeric> <integer> <integer> <integer>
## [1] 33 NaN 1 0 1
## [2] 35.375 NaN 6 0 2
## [3] 35.75 NaN 0 0 4
## [4] 34.75 NaN 1 0 3
## count.minus.ref read.pos.mean read.pos.mean.ref read.pos.var
## <integer> <numeric> <numeric> <numeric>
## [1] 0 36.5 NaN 2112.5
## [2] 0 64.125 NaN 990.984375
## [3] 0 49.75 NaN 2435.9375
## [4] 0 31.5 NaN 1363.75
## read.pos.var.ref mdfne mdfne.ref
## <numeric> <numeric> <numeric>
## [1] <NA> <NA> <NA>
## [2] <NA> <NA> <NA>
## [3] <NA> <NA> <NA>
## [4] <NA> <NA> <NA>
## ---
## seqlengths:
## TP53
## 2025772
## hardFilters(4): nonRef nonNRef readCount likelihoodRatio
Now we will call variants for the whole region:
tally.param <- TallyVariantsParam(genome, high_base_quality = 23L)
called.variants <- callVariants(bam, tally.param)
called.variants
## VRanges with 45 ranges and 15 metadata columns:
## seqnames ranges strand ref alt
## <Rle> <IRanges> <Rle> <character> <characterOrRle>
## [1] TP53 [352607, 352607] + C T
## [2] TP53 [352702, 352702] + T C
## [3] TP53 [353997, 353997] + T C
## [4] TP53 [589438, 589438] + G A
## [5] TP53 [589486, 589486] + T C
## ... ... ... ... ... ...
## [41] TP53 [1813169, 1813169] + T C
## [42] TP53 [1813170, 1813170] + G A
## [43] TP53 [1813836, 1813836] + A G
## [44] TP53 [1851805, 1851805] + C T
## [45] TP53 [1859174, 1859174] + G A
## totalDepth refDepth altDepth sampleNames
## <integerOrRle> <integerOrRle> <integerOrRle> <factorOrRle>
## [1] 10 0 10 <NA>
## [2] 4 0 4 <NA>
## [3] 3 0 3 <NA>
## [4] 4 0 4 <NA>
## [5] 3 0 3 <NA>
## ... ... ... ... ...
## [41] 3 0 3 <NA>
## [42] 3 0 3 <NA>
## [43] 7 0 7 <NA>
## [44] 2 0 2 <NA>
## [45] 2 0 2 <NA>
## softFilterMatrix | raw.count raw.count.ref raw.count.total
## <matrix> | <integer> <integer> <integer>
## [1] | 10 0 10
## [2] | 4 0 4
## [3] | 3 0 3
## [4] | 4 0 4
## [5] | 3 0 3
## ... ... ... ... ... ...
## [41] | 3 0 3
## [42] | 3 0 3
## [43] | 7 0 7
## [44] | 2 0 2
## [45] | 2 0 2
## mean.quality mean.quality.ref count.plus count.plus.ref
## <numeric> <numeric> <integer> <integer>
## [1] 34.3 NaN 2 0
## [2] 34.75 NaN 0 0
## [3] 29.6666666666667 NaN 2 0
## [4] 34.5 NaN 1 0
## [5] 38 NaN 1 0
## ... ... ... ... ...
## [41] 25 NaN 1 0
## [42] 24 NaN 1 0
## [43] 38.1428571428571 NaN 3 0
## [44] 39 NaN 0 0
## [45] 37 NaN 1 0
## count.minus count.minus.ref read.pos.mean read.pos.mean.ref
## <integer> <integer> <numeric> <numeric>
## [1] 8 0 62.9 NaN
## [2] 4 0 27.25 NaN
## [3] 1 0 62.3333333333333 NaN
## [4] 3 0 45 NaN
## [5] 2 0 36.3333333333333 NaN
## ... ... ... ... ...
## [41] 2 0 75.3333333333333 NaN
## [42] 2 0 75 NaN
## [43] 4 0 30.7142857142857 NaN
## [44] 2 0 31.5 NaN
## [45] 1 0 62.5 NaN
## read.pos.var read.pos.var.ref mdfne mdfne.ref
## <numeric> <numeric> <numeric> <numeric>
## [1] 1106.36777777778 <NA> <NA> <NA>
## [2] 421.770833333333 <NA> <NA> <NA>
## [3] 3255.05555555556 <NA> <NA> <NA>
## [4] 1881 <NA> <NA> <NA>
## [5] 962.388888888889 <NA> <NA> <NA>
## ... ... ... ... ...
## [41] 2997.88888888889 <NA> <NA> <NA>
## [42] 2969.5 <NA> <NA> <NA>
## [43] 1263.46598639456 <NA> <NA> <NA>
## [44] 924.5 <NA> <NA> <NA>
## [45] 2112.5 <NA> <NA> <NA>
## ---
## seqlengths:
## TP53
## 2025772
## hardFilters(4): nonRef nonNRef readCount likelihoodRatio
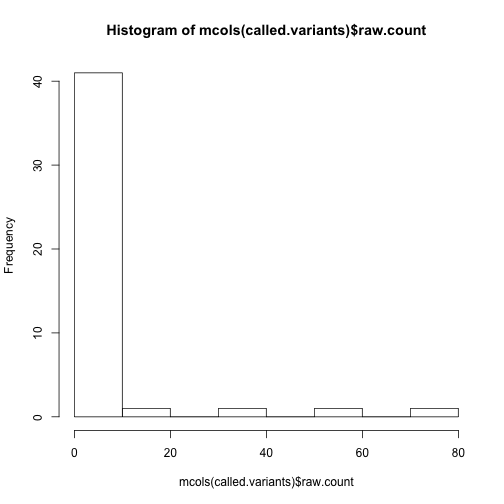
How many non-reference alleles do we see:
hist(mcols(called.variants)$raw.count)
with(mcols(called.variants), plot(raw.count.total, raw.count, log = "xy"))
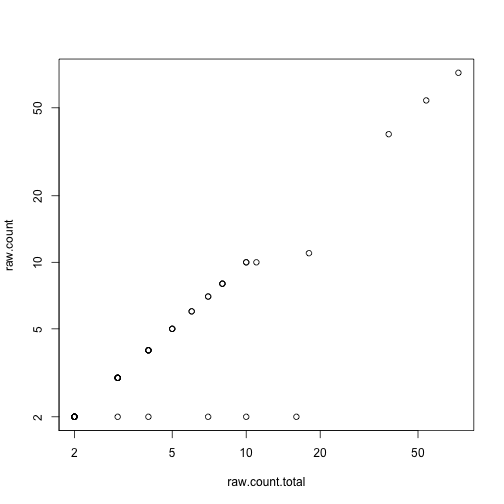
(i <- which.max(mcols(called.variants)$raw.count))
## [1] 9
called.variants[i, ]
## VRanges with 1 range and 15 metadata columns:
## seqnames ranges strand ref alt
## <Rle> <IRanges> <Rle> <character> <characterOrRle>
## [1] TP53 [650046, 650046] + G A
## totalDepth refDepth altDepth sampleNames
## <integerOrRle> <integerOrRle> <integerOrRle> <factorOrRle>
## [1] 67 0 67 <NA>
## softFilterMatrix | raw.count raw.count.ref raw.count.total
## <matrix> | <integer> <integer> <integer>
## [1] | 72 1 73
## mean.quality mean.quality.ref count.plus count.plus.ref
## <numeric> <numeric> <integer> <integer>
## [1] 36.1492537313433 NaN 43 0
## count.minus count.minus.ref read.pos.mean read.pos.mean.ref
## <integer> <integer> <numeric> <numeric>
## [1] 29 1 51 85
## read.pos.var read.pos.var.ref mdfne mdfne.ref
## <numeric> <numeric> <numeric> <numeric>
## [1] 580.464788732394 <NA> <NA> <NA>
## ---
## seqlengths:
## TP53
## 2025772
## hardFilters(4): nonRef nonNRef readCount likelihoodRatio
Writing out VCF files
sampleNames(called.variants) <- "H1993"
mcols(called.variants) <- NULL
vcf <- asVCF(called.variants)
writeVcf(vcf, "H1993.vcf", index = TRUE)
A few more details:
# ?postFilterVariants ?callSampleSpecificVariants
somatic <- callSampleSpecificVariants(bams$H1993, bams$H2073, tally.param)
Reading in VCF and annotating
To annotate the VCF, we start by loading a UCSC transcript database:
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
Now we read in a small subset of a VCF file, over chr22, which is stored in the VariantAnnotation package. Note that the readVcf function can be slow if the file is many Mb. This one is less than 1 Mb.
library(VariantAnnotation)
fl <- system.file("extdata", "chr22.vcf.gz", package = "VariantAnnotation")
vcf <- readVcf(fl, genome = "hg19")
rowData(vcf)
## GRanges with 10376 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## rs7410291 22 [50300078, 50300078] * | <NA>
## rs147922003 22 [50300086, 50300086] * | <NA>
## rs114143073 22 [50300101, 50300101] * | <NA>
## rs141778433 22 [50300113, 50300113] * | <NA>
## rs182170314 22 [50300166, 50300166] * | <NA>
## ... ... ... ... ... ...
## rs187302552 22 [50999536, 50999536] * | <NA>
## rs9628178 22 [50999538, 50999538] * | <NA>
## rs5770892 22 [50999681, 50999681] * | <NA>
## rs144055359 22 [50999830, 50999830] * | <NA>
## rs114526001 22 [50999964, 50999964] * | <NA>
## REF ALT QUAL FILTER
## <DNAStringSet> <DNAStringSetList> <numeric> <character>
## rs7410291 A G 100 PASS
## rs147922003 C T 100 PASS
## rs114143073 G A 100 PASS
## rs141778433 C T 100 PASS
## rs182170314 C T 100 PASS
## ... ... ... ... ...
## rs187302552 A G 100 PASS
## rs9628178 A G 100 PASS
## rs5770892 A G 100 PASS
## rs144055359 G A 100 PASS
## rs114526001 G C 100 PASS
## ---
## seqlengths:
## 22
## NA
Note that we can also restrict the amount we read in, either by specifying a range, or using yieldSize
param <- ScanVcfParam(which = GRanges("22", IRanges(50500000, 50600000)))
vcf <- readVcf(fl, genome = "hg19", param = param)
rowData(vcf)
## GRanges with 1726 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## rs142027672 22 [50500000, 50500000] * | <NA>
## 22:50500032_C/T 22 [50500032, 50500032] * | <NA>
## rs75152548 22 [50500263, 50500263] * | <NA>
## rs190746868 22 [50500297, 50500297] * | <NA>
## rs183916737 22 [50500375, 50500375] * | <NA>
## ... ... ... ... ... ...
## rs187838910 22 [50599491, 50599491] * | <NA>
## rs148213119 22 [50599735, 50599735] * | <NA>
## rs141267497 22 [50599771, 50599771] * | <NA>
## 22:50599777_C/T 22 [50599777, 50599777] * | <NA>
## rs150761060 22 [50599930, 50599930] * | <NA>
## REF ALT QUAL FILTER
## <DNAStringSet> <DNAStringSetList> <numeric> <character>
## rs142027672 C T 100 PASS
## 22:50500032_C/T C T 100 PASS
## rs75152548 T C 100 PASS
## rs190746868 C T 100 PASS
## rs183916737 G T 100 PASS
## ... ... ... ... ...
## rs187838910 G A 100 PASS
## rs148213119 G A 100 PASS
## rs141267497 G A 100 PASS
## 22:50599777_C/T C T 100 PASS
## rs150761060 C T 100 PASS
## ---
## seqlengths:
## 22
## NA
# ?readVcf
We can find which variants overlap coding regions with the locateVariants function:
# here we hack the chromosome from '22' to 'chr22'
seqlevels(vcf) <- paste0("chr", seqlevels(vcf))
loc <- locateVariants(vcf, txdb, CodingVariants())
loc
## GRanges with 455 ranges and 7 metadata columns:
## seqnames ranges strand | LOCATION QUERYID
## <Rle> <IRanges> <Rle> | <factor> <integer>
## [1] chr22 [50500032, 50500032] - | coding 2
## [2] chr22 [50500032, 50500032] - | coding 2
## [3] chr22 [50500032, 50500032] - | coding 2
## [4] chr22 [50500032, 50500032] - | coding 2
## [5] chr22 [50500032, 50500032] - | coding 2
## ... ... ... ... ... ... ...
## [451] chr22 [50599253, 50599253] + | coding 1717
## [452] chr22 [50599253, 50599253] + | coding 1717
## [453] chr22 [50599466, 50599466] + | coding 1721
## [454] chr22 [50599466, 50599466] + | coding 1721
## [455] chr22 [50599466, 50599466] + | coding 1721
## TXID CDSID GENEID PRECEDEID FOLLOWID
## <integer> <integer> <character> <CharacterList> <CharacterList>
## [1] 75255 218579 23209
## [2] 75256 218579 23209
## [3] 75257 218579 23209
## [4] 75258 218579 23209
## [5] 75259 218579 23209
## ... ... ... ... ... ...
## [451] 74369 216346 54456
## [452] 74372 216346 54456
## [453] 74366 216347 54456
## [454] 74367 216347 54456
## [455] 74372 216347 54456
## ---
## seqlengths:
## chr22
## NA
We can double check that it did what we expected:
loc[1]
## GRanges with 1 range and 7 metadata columns:
## seqnames ranges strand | LOCATION QUERYID TXID
## <Rle> <IRanges> <Rle> | <factor> <integer> <integer>
## [1] chr22 [50500032, 50500032] - | coding 2 75255
## CDSID GENEID PRECEDEID FOLLOWID
## <integer> <character> <CharacterList> <CharacterList>
## [1] 218579 23209
## ---
## seqlengths:
## chr22
## NA
g <- genes(txdb)
fo <- findOverlaps(loc[1], g)
fo
## Hits of length 1
## queryLength: 1
## subjectLength: 23056
## queryHits subjectHits
## <integer> <integer>
## 1 1 7061
g[subjectHits(fo)]
## GRanges with 1 range and 1 metadata column:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <CharacterList>
## 23209 chr22 [50497820, 50524358] - | 23209
## ---
## seqlengths:
## chr1 chr2 ... chrUn_gl000249
## 249250621 243199373 ... 38502
loc[1]
## GRanges with 1 range and 7 metadata columns:
## seqnames ranges strand | LOCATION QUERYID TXID
## <Rle> <IRanges> <Rle> | <factor> <integer> <integer>
## [1] chr22 [50500032, 50500032] - | coding 2 75255
## CDSID GENEID PRECEDEID FOLLOWID
## <integer> <character> <CharacterList> <CharacterList>
## [1] 218579 23209
## ---
## seqlengths:
## chr22
## NA