SummarizedExperiment class in depth
Introduction
In the data management overview, we applied summarizeOverlaps to
the set of BAM files in RNAseqData.HNRNPC.bam.chr14. Let’s do it
again to focus on the object that is returned.
BAM files to SummarizedExperiment for a single region
library(RNAseqData.HNRNPC.bam.chr14)
bfp = RNAseqData.HNRNPC.bam.chr14_BAMFILES
library(Rsamtools)
bfl = BamFileList(file=bfp)
hnrnpcLoc = GRanges("chr14", IRanges(21677296, 21737638))
library(GenomicAlignments)
library(BiocParallel)
register(SerialParam())
hnse = summarizeOverlaps(hnrnpcLoc,bfl)
hnse
## class: RangedSummarizedExperiment
## dim: 1 8
## metadata(0):
## assays(1): counts
## rownames: NULL
## rowData names(0):
## colnames(8): ERR127306 ERR127307 ... ERR127304 ERR127305
## colData names(0):
hnse is an instance of RangedSummarizedExperiment. This class is like
an ExpressionSet but has more facilities (and more modern
facilities) for managing assay summaries and metadata.
A visual schematic follows:
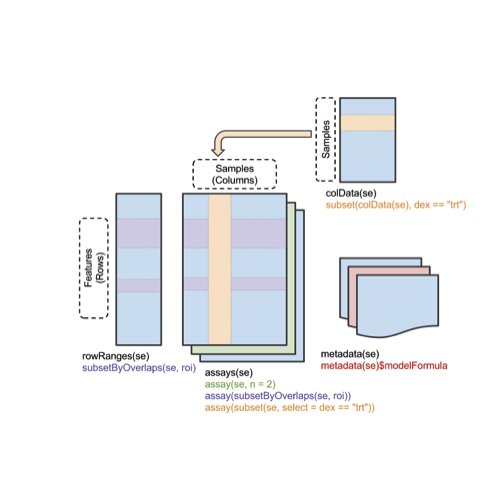
Effective use of SummarizedExperiment instances involves learning
about methods that have been defined for them.
In order to get at the read/region overlap counts for HNRNPC, we apply
the assay method:
assay(hnse)
## ERR127306 ERR127307 ERR127308 ERR127309 ERR127302 ERR127303 ERR127304
## [1,] 5422 6320 5896 5558 172 196 316
## ERR127305
## [1,] 282
This is a bare-bones representation of the result. The sample identifiers have been propagated to column names of the matrix of counts, but information on the region examined is lost in this display.
Metadata opportunities in the SummarizedExperiment
The hnse object has a little more information.
rowRanges(hnse)
## GRanges object with 1 range and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr14 [21677296, 21737638] *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
seqinfo(hnse)
## Seqinfo object with 1 sequence from an unspecified genome; no seqlengths:
## seqnames seqlengths isCircular genome
## chr14 NA NA <NA>
metadata(hnse)
## list()
We can do better. We will set up the analysis differently so that the output is more comprehensive and self-describing.
Making SummarizedExperiment more effective by enriching the inputs to summarizing methods
Defining regions of interest, with metadata
We have seen that it is sufficient to define a single GRanges
to drive summarizeOverlaps over a set of BAM files. We’d
like to preserve more metadata about the regions examined.
We’ll use the TxDb infrastructure, to be described in more detail
later, to get a structure defining gene regions on chr14.
We’ll also use the Homo.sapiens annotation package to add
gene symbols.
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb = TxDb.Hsapiens.UCSC.hg19.knownGene
gr14 = genes(txdb, vals=list(tx_chrom="chr14"))
## Warning: 'elementLengths' is deprecated.
## Use 'elementNROWS' instead.
## See help("Deprecated")
## Warning: 'elementLengths' is deprecated.
## Use 'elementNROWS' instead.
## See help("Deprecated")
gr14$symbol = mapIds(Homo.sapiens, keys=gr14$gene_id, keytype="ENTREZID",
column="SYMBOL")
## 'select()' returned 1:1 mapping between keys and columns
gr14
## GRanges object with 781 ranges and 2 metadata columns:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <character>
## 10001 chr14 [ 71050957, 71067384] - | 10001
## 100113389 chr14 [ 45580078, 45580176] + | 100113389
## 100113391 chr14 [ 20794600, 20794698] - | 100113391
## 100124539 chr14 [ 91592770, 91592896] + | 100124539
## 100126297 chr14 [101507700, 101507781] + | 100126297
## ... ... ... ... . ...
## 9870 chr14 [ 75127955, 75179807] - | 9870
## 9878 chr14 [ 21945335, 21967319] + | 9878
## 9895 chr14 [102829300, 102968818] + | 9895
## 9950 chr14 [ 93260650, 93306304] + | 9950
## 9985 chr14 [ 24641234, 24649463] + | 9985
## symbol
## <character>
## 10001 MED6
## 100113389 SNORD127
## 100113391 SNORD126
## 100124539 SNORA11B
## 100126297 MIR300
## ... ...
## 9870 AREL1
## 9878 TOX4
## 9895 TECPR2
## 9950 GOLGA5
## 9985 REC8
## -------
## seqinfo: 93 sequences (1 circular) from hg19 genome
Defining the sample characteristics for the BAM files
We have three distinct preparations, one for control and two for knockdown. We will use the GenomicFiles infrastructure to bind the sample level metadata.
char = rep(c("hela_wt", "hela_hkd"), each=4)
bff = GenomicFiles(files=path(bfl))
colData(bff)$condition = char
sid = c(1,1,1,1,2,2,3,3)
bff$sample = sid
bff
## GenomicFiles object with 0 ranges and 8 files:
## files: ERR127306_chr14.bam, ERR127307_chr14.bam, ..., ERR127304_chr14.bam, ERR127305_chr14.bam
## detail: use files(), rowRanges(), colData(), ...
Comparing read overlaps, preserving metadata
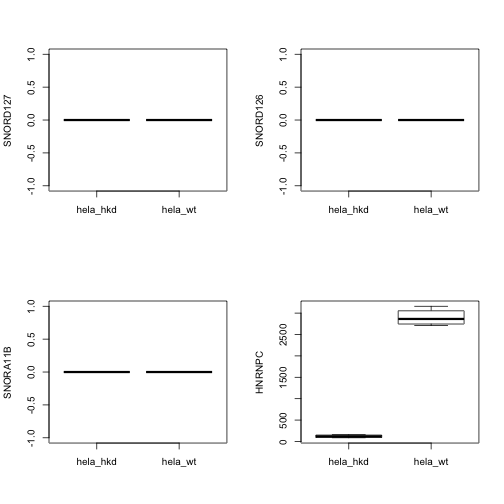
We’ll look at 5 genes, including HNRNPC. After computing, we bind the sample-level data back into the result.
hnse = summarizeOverlaps(gr14[c(1:4,305)],files(bff))
colData(hnse) = cbind(colData(hnse), colData(bff))
hnse
## class: RangedSummarizedExperiment
## dim: 5 8
## metadata(0):
## assays(1): counts
## rownames(5): 10001 100113389 100113391 100124539 3183
## rowData names(2): gene_id symbol
## colnames(8): ERR127306 ERR127307 ... ERR127304 ERR127305
## colData names(2): condition sample
assay(hnse)
## ERR127306 ERR127307 ERR127308 ERR127309 ERR127302 ERR127303
## 10001 114 156 129 144 175 213
## 100113389 0 0 0 0 0 0
## 100113391 0 0 0 0 0 0
## 100124539 0 0 0 0 0 0
## 3183 2711 3160 2948 2779 86 98
## ERR127304 ERR127305
## 10001 210 165
## 100113389 0 0
## 100113391 0 0
## 100124539 0 0
## 3183 158 141
Note that row identifiers are now present with the count matrix.
A simple sanity check:
par(mfrow=c(2,2))
for (i in 2:5) {
boxplot(assay(hnse)[i,]~hnse$condition, ylab=rowRanges(hnse)$symbol[i])
}
Converting from ExpressionSet
This is easy, but more work will be needed to allow subsetting of array probes based on genomic range queries.
library(ALL)
data(ALL)
allse = makeSummarizedExperimentFromExpressionSet(ALL)
allse
## class: RangedSummarizedExperiment
## dim: 12625 128
## metadata(3): experimentData annotation protocolData
## assays(1): exprs
## rownames(12625): 1000_at 1001_at ... AFFX-YEL021w/URA3_at
## AFFX-YEL024w/RIP1_at
## rowData names(0):
## colnames(128): 01005 01010 ... 83001 LAL4
## colData names(21): cod diagnosis ... f.u date.last.seen
rowRanges(allse)
## Warning: 'elementLengths' is deprecated.
## Use 'elementNROWS' instead.
## See help("Deprecated")
## GRangesList object of length 12625:
## $$1000_at
## GRanges object with 0 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
##
## $$1001_at
## GRanges object with 0 ranges and 0 metadata columns:
## seqnames ranges strand
##
## $$1002_f_at
## GRanges object with 0 ranges and 0 metadata columns:
## seqnames ranges strand
##
## ...
## <12622 more elements>
## -------
## seqinfo: no sequences
Summary
The RangedSummarizedExperiment class instantiates some of the key principles of Bioconductor data structure design:
- Assay data and metadata on sample characteristics (colData) are bound together in a coordinated way
- Matrix-like subsetting works directly on both assay and sample data
- Range-based subsetting works for assay components addressible by genomic coordinates
- Arbitrary metadata on assay features can be provided in the mcols(rowRanges(se))
- Arbitrary general metadata can be provided through
metadata(se)<-
We’ll learn more about adaptations of SummarizedExperiment to perform specifically for multistage processing and analysis of RNA-seq experiments later in the course.