Understanding and building R packages
What is an R package?
Conceptually, an R package is a collection of functions, data objects, and documentation that coherently support a family of related data analysis operations.
Concretely, an R package is a structured collection of folders, organized and populated according to the rules of Writing R Extensions.
A new software package with package.skeleton
We can create our own packages using package.skeleton. We’ll illustrate that now
with an enhancement to the ERBS package that was created for the course.
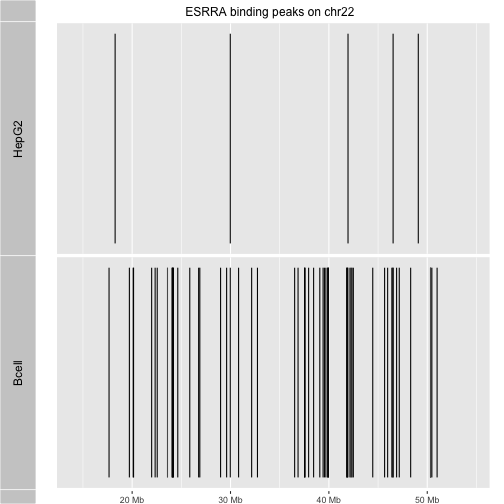
We’ll create a new package that utilizes the peak data, defining
a function juxta that allows us to compare binding peak patterns for the two cell
types on a selected chromosome. (I have commented out code that
uses an alternative graphics engine, for optional exploration.)
Here’s a definition of juxta. Add it to your R session.
juxta = function (chrname="chr22", ...)
{
require(ERBS)
data(HepG2)
data(GM12878)
require(ggbio)
require(GenomicRanges) # "subset" is overused, need import detail
ap1 = autoplot(HepG2[which(seqnames(HepG2)==chrname)])
ap2 = autoplot(GM12878[which(seqnames(GM12878)==chrname)])
tracks(HepG2 = ap1, Bcell = ap2, ...)
# alternative code for Gviz below
# require(Gviz)
# names(ap1) = "HepG2"
# names(ap2) = "B-cell"
# ax = GenomeAxisTrack()
# plotTracks(list(ax, ap1, ap2))
}
Now demonstrate it as follows.
library(ERBS)
juxta("chr22", main="ESRRA binding peaks on chr22")
In the video we will show how to use package.skeleton and the Rstudio
editor to generate, document, and install this new package! We will not
streamline the code in juxta to make use of inter-package
symbol transfer by properly writing the DESCRIPTION and NAMESPACE
files for the package, but leave this for an advanced course in
software development.
A new annotation package with OrganismDbi
We have found the Homo.sapiens package to be quite convenient.
We can get gene models, symbol to GO mappings, and so on, without
remembering any more than keytypes, columns, keys, and select.
At present there is no similar resource for S. cerevisiae.
We can make one, following the OrganismDbi vignette. This is
a very lightweight integrative package.
library(OrganismDbi)
gd = list( join1 = c(GO.db="GOID", org.Sc.sgd.db="GO"),
join2 = c(org.Sc.sgd.db="ENTREZID",
TxDb.Scerevisiae.UCSC.sacCer3.sgdGene="GENEID"))
if (!file.exists("Sac.cer3")) # don't do twice...
makeOrganismPackage(pkgname="Sac.cer3", # simplify typing!
graphData=gd, organism="Saccharomyces cerevisiae",
version="1.0.0", maintainer="Student <ph525x@harvardx.edu>",
author="Student <ph525x@harvardx.edu>",
destDir=".",
license="Artistic-2.0")
At this point we have a folder structure in our working folder that can support an installation.
install.packages("Sac.cer3", repos=NULL, type="source")
library(Sac.cer3)
## Loading required package: org.Sc.sgd.db
##
Sac.cer3
## OrganismDb Object:
## # Includes GODb Object: GO.db
## # With data about: Gene Ontology
## # Includes OrgDb Object: org.Sc.sgd.db
## # Gene data about: Saccharomyces cerevisiae
## # Taxonomy Id: 559292
## # Includes TxDb Object: TxDb.Scerevisiae.UCSC.sacCer3.sgdGene
## # Transcriptome data about: Saccharomyces cerevisiae
## # Based on genome: sacCer3
## # The OrgDb gene id ENTREZID is mapped to the TxDb gene id GENEID .
columns(Sac.cer3)
## [1] "ALIAS" "CDSCHROM" "CDSEND" "CDSID"
## [5] "CDSNAME" "CDSSTART" "CDSSTRAND" "COMMON"
## [9] "DEFINITION" "DESCRIPTION" "ENSEMBL" "ENSEMBLPROT"
## [13] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
## [17] "EVIDENCEALL" "EXONCHROM" "EXONEND" "EXONID"
## [21] "EXONNAME" "EXONRANK" "EXONSTART" "EXONSTRAND"
## [25] "GENEID" "GENENAME" "GO" "GOALL"
## [29] "GOID" "INTERPRO" "ONTOLOGY" "ONTOLOGYALL"
## [33] "ORF" "PATH" "PFAM" "PMID"
## [37] "REFSEQ" "SGD" "SMART" "TERM"
## [41] "TXCHROM" "TXEND" "TXID" "TXNAME"
## [45] "TXSTART" "TXSTRAND" "TXTYPE" "UNIPROT"
genes(Sac.cer3)
## GRanges object with 6534 ranges and 1 metadata column:
## seqnames ranges strand | GENEID
## <Rle> <IRanges> <Rle> | <FactorList>
## Q0010 chrM [ 3952, 4415] + | Q0010
## Q0032 chrM [11667, 11957] + | Q0032
## Q0055 chrM [13818, 26701] + | Q0055
## Q0075 chrM [24156, 25255] + | Q0075
## Q0080 chrM [27666, 27812] + | Q0080
## ... ... ... ... . ...
## YPR200C chrXVI [939279, 939671] - | YPR200C
## YPR201W chrXVI [939922, 941136] + | YPR201W
## YPR202W chrXVI [943032, 944188] + | YPR202W
## YPR204C-A chrXVI [946856, 947338] - | YPR204C-A
## YPR204W chrXVI [944603, 947701] + | YPR204W
## -------
## seqinfo: 17 sequences (1 circular) from sacCer3 genome
Using devtools
Let’s use devtools to create a package centered
around the juxta function. We will learn about
roxygen too.
In the following code chunk, we use devtools::create
to generate a package folder structure in a temporary
directory.
curd = getwd()
kk = dir.create(tmpd <- tempfile()) # always new
setwd(tmpd)
library(devtools)
create("juxtaPack", list(Depends=c("ERBS", "ggbio"),
Description="Juxtapose ESRRA binding and gene models",
Title="demonstration package"))
## Package: juxtaPack
## Title: demonstration package
## Version: 0.0.0.9000
## Authors@R: person("First", "Last", email = "first.last@example.com", role = c("aut", "cre"))
## Description: Juxtapose ESRRA binding and gene models
## Depends: ERBS, ggbio
## License: What license is it under?
## Encoding: UTF-8
## LazyData: true
setwd("juxtaPack")
We are now in the top folder of the package hierarchy.
We will create a text file with documentation (prefixed
by hashtag single quote) in the roxygen2 format.
The text vector jlines defines the content. (Usually
you will create your documentation and function code
in a text editor.)
setwd(tmpd)
setwd("juxtaPack")
jlines =
"#' render a chromosome and locations of ESRRA binding sites
#' @param chrname character(1) giving UCSC chromosome name
#' @examples
#' juxta()
#' @export
juxta = function (chrname='chr22', ...)
{
require(ERBS)
data(HepG2)
data(GM12878)
require(ggbio)
require(GenomicRanges)
ac = as.character
ap1 = autoplot(HepG2[which(ac(seqnames(HepG2))==chrname)])
ap2 = autoplot(GM12878[which(ac(seqnames(GM12878))==chrname)])
tracks(HepG2 = ap1, Bcell = ap2, ...)
}
"
We descend to the R folder and write our text file.
setwd(tmpd)
setwd("juxtaPack")
setwd("R")
writeLines(jlines, "jux.R")
We ascend to the package root and run document and install
to get access to the new package.
setwd(tmpd)
setwd("juxtaPack")
document()
## Updating juxtaPack documentation
## Loading juxtaPack
## Updating roxygen version in /private/var/folders/5_/14ld0y7s0vbg_z0g2c9l8v300000gr/T/Rtmprv6lu6/file99ae46a8f0f5/juxtaPack/DESCRIPTION
## Writing NAMESPACE
## Writing juxta.Rd
install()
## Installing juxtaPack
## '/Library/Frameworks/R.framework/Resources/bin/R' --no-site-file \
## --no-environ --no-save --no-restore --quiet CMD INSTALL \
## '/private/var/folders/5_/14ld0y7s0vbg_z0g2c9l8v300000gr/T/Rtmprv6lu6/file99ae46a8f0f5/juxtaPack' \
## --library='/Library/Frameworks/R.framework/Versions/3.4/Resources/library' \
## --install-tests
##
## Reloading installed juxtaPack
##
## Attaching package: 'juxtaPack'
## The following object is masked _by_ '.GlobalEnv':
##
## juxta
setwd(curd) # go back to where you started
library(juxtaPack)
juxta
## function (chrname="chr22", ...)
## {
## require(ERBS)
## data(HepG2)
## data(GM12878)
## require(ggbio)
## require(GenomicRanges) # "subset" is overused, need import detail
## ap1 = autoplot(HepG2[which(seqnames(HepG2)==chrname)])
## ap2 = autoplot(GM12878[which(seqnames(GM12878)==chrname)])
## tracks(HepG2 = ap1, Bcell = ap2, ...)
## # alternative code for Gviz below
## # require(Gviz)
## # names(ap1) = "HepG2"
## # names(ap2) = "B-cell"
## # ax = GenomeAxisTrack()
## # plotTracks(list(ax, ap1, ap2))
## }
In summary:
- We have used
writeLinesto generate a combination of roxygen documentation and R code in the filejux.R, in folderR, which was created as a subfolder of folderjuxtaPack. - We then changed to the
juxtaPackfolder and randocument()that will translate the roxygen lines to statements inNAMESPACEand to a .Rd file inman. - We then ran
install()to install the package in R, returned to our initial folder, and usedlibrary(juxtaPack)to attach our new package.
Full development would include production of a vignette
and a suite of unit tests, giving
a meaningful basis for using check() in devtools.
In a more extensive course,
these would be addressed, but you can learn about them yourself
by looking at any Bioconductor package. A good example is
IRanges, which has extensive unit testing.
There are many other good examples.
Wrapping up
You are now in a good position to revisit the motivations and core values section of the course pages.
In that section we described the concepts of functional object-oriented programming and continuous integration as they pertain to the development of highly reliable and relevant software tools, used in a variety of subdomains of genome science. The software engineering and reproducible research underpinnings of the Bioconductor project are at the heart of its scientific impact. For a nice set of remarks on usability in upcoming cloud contexts, see this recent Nature Toolbox commentary.